- Collection of ChIP-chip/ChIP-seq datasets of histone modifications, histone regulators and chromatin regulators
- Defining genes whose promoters/coding regions contain a specific histone modification
- Defining genes whose promoters are bound by a specific histone/chromatin regulator
- Identifying the enriched/depleted histone modifications and the enriched histone/chromatin regulators for the user's input genes
- Implementation and maintenance of the web interface of YHMI
- Download the command line program of YHMI
About YHMI
Post-translational modifications of histones (e.g. acetylation, methylation, phosphorylation, and ubiquitination) play crucial roles in regulating gene expression by altering chromatin structures and creating docking sites for histone/chromatin regulators. However, the combination patterns of histone modifications, regulatory proteins, and their corresponding target genes remain incompletely understood. Therefore, it is advantageous to have a tool for the enrichment/depletion analysis of histone modifications and histone/chromatin regulators from a gene list. Many ChIP-chip/ChIP-seq datasets of histone modifications and histone/chromatin regulators in yeast can be found in the literature. Knowing the needs and having the data motivate us to develop a web tool, called YHMI (Yeast Histone Modifications Identifier), which can identify the enriched/depleted histone modifications and the enriched histone/chromatin regulators from a list of yeast genes.
Here we develop a web tool called YHMI (Yeast Histone Modification Identifier). YHMI uses ChIP-chip/ChIP-seq datasets of 32 histone modifications (15 histone acetylation, 13 histone methylation, 2 histone phosphorylation, 1 histone ubiquitination, and 1 histone variant) and 83 histone/chromatin regulators (18-24). When a user inputs a gene list, YHMI will identify the enriched/depleted histone modifications in the promoters/coding regions and the enriched histone/chromatin regulators in the promoters of the genes in the input list. The identification results are shown both in tables and figures. Therefore, YHMI can be used to shed light on what is unknown in a gene list of interest. Several possible biological questions could be answered by YHMI. For example, what are the enriched/depleted histone codes in a gene list of a specific property (e.g. highly transcribed genes, stress-responsive genes, and genes in a specific pathway)? What are the enriched/depleted histone codes in a gene list associated with a specific factor (e.g. target genes of a transcription factor, lipid-binding proteins, and hexose transporter genes)?
Construction of YHMI
All the ChIP-chip/ChIP-seq data used in YHMI were downloaded from SGD. SGD collected the raw data of ChIP-chip/ChIP-seq from GEO and ArrayExpress. Then SGD mapped these data to the latest yeast reference genome sacCer3 (R64) and allowed everyone to download the processed data. Therefore, we directly downloaded the ChIP-chip/ChIP-seq datasets of 32 histone modifications and 83 histone/chromatin regulators from SGD.
Table 1.
| 15 histone acetylation | H2AK5ac, H3K4ac, H3K9ac, H3K14ac, H3K14ac [H2O2], H3K18ac, H3K23ac, H3K27ac, H3K56ac, H4ac, H4ac [H2O2], H4K5ac, H4K8ac, H4K12ac, H4K16ac |
| 13 histone methylation | H3R2me2a, H3K4me, H3K4me2, H3K4me3, H3K36me, H3K36me2, H3K36me3, H3K79me, H3K79me2, H3K79me3, H4R3me, H4R3me2s, H4K20me |
| 2 histone phosphorylation | H2AS129ph, H3S10ph |
| 1 histone ubiquitination | H2BK123ub |
| 1 histone variant | H2AZ |
| 83 histone/chromatin regulators (in 25℃ and 37℃) | Ada2, Brn1, Esa1, Gcn5, Rsc4, Rxt2, Snf1, Swc1, Yaf9, ... See more. |
A gene's promoter is defined as the region between 500 bp upstream and 100 bp downstream of the start codon. A gene's coding region is defined as the region between the start codon and the stop codon. The procedure of defining a set of genes whose promoters/coding regions contain a specific histone modification (e.g. H3K4ac) is as follows.
- For each of the 6572 genes in the yeast genome, we extracted the maximal data value (log2(H3K4ac/H3) in this case) in its promoter/coding region from the corresponding ChIP-chip dataset.
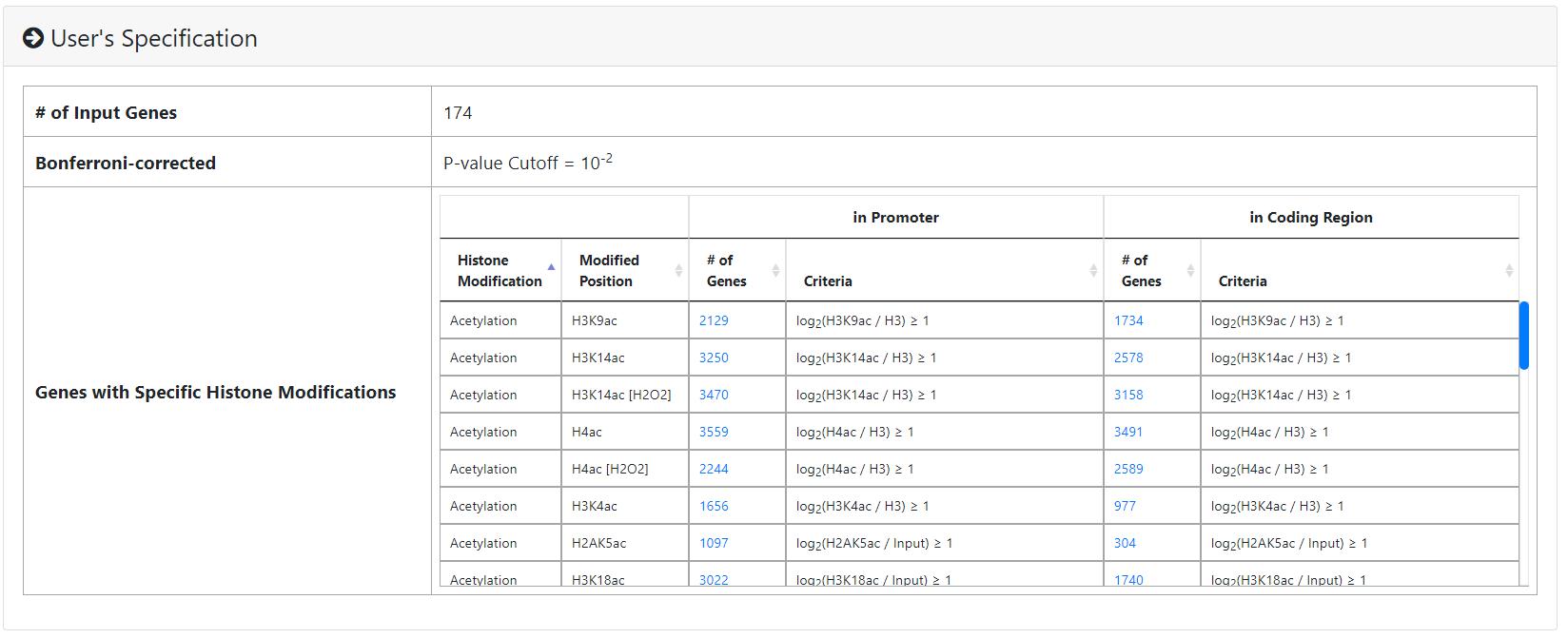
- A gene whose promoter/coding region is said to contain H3K4ac if its promoter/coding region satisfies log2(H3K4ac/H3)≥threshold, where the threshold is set by the user. For example, 1656 genes' promoters and 977 gene's coding regions are said to contain H3K4ac when 1 is set as the threshold.
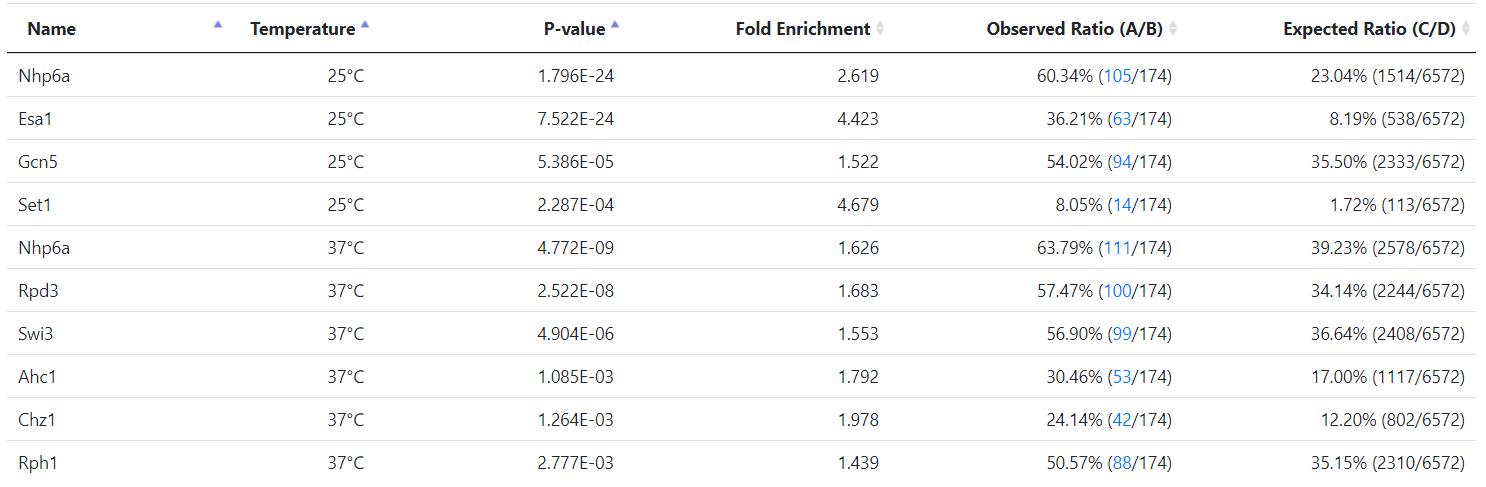
Venters et al. identified high-confident (less than 5% FDR) interactions between a specific histone/chromatin regulator and genomic DNA (in the yeast genome) under normal (25℃) and acute heat-shock (37℃) conditions by ChIP-chip experiments. Using Venter et al.'s results and based on the definition of a gene's promoter region, we can determine the genes whose promoters are bound by a histone/chromatin regulator. Supplementary Table 1 provides the number and the names of genes whose promoters are bound by the 83 histone/chromatin regulators under normal (25℃) and acute heat-shock (37℃) conditions.
The main functionality of YHMI is to identify the enriched/depleted histone modifications and the enriched histone/chromatin regulators for the user's input genes. The hypergeometric distribution is used to test the statistical significance of enrichment. The procedure for checking whether a specific histone modification (e.g. H3K4ac) is enriched/depleted in the promoters of the user's input genes is as follows.
- Let \(S\) be the set of genes whose promoters contain the histone modification H3K4ac, \(R\) be the set of the user's input genes, \(T=S∩R\) be the set of genes whose promoters contain H3K4ac and are also in the set of the user's input genes, and \(F\) be the set of all genes in the yeast genome.
-
The \(p_{\_value}(enrichment)\) and \(p_{\_value}(depletion)\) for rejecting the null hypothesis (H0: H3K4ac is not enriched/depleted in the promoters of the user's input genes) are calculated as
$$ p_{\_value}(enrichment) = P(x≥|T|) = \sum^{min(|S|,|R|)}_{x=|T|} \frac{(\begin{matrix}|S|\\x\end{matrix})(\begin{matrix}|F|-|S|\\|R|-x\end{matrix})}{(\begin{matrix}|F|\\|R|\end{matrix})}\qquad$$
$$ p_{\_value}(depletion) = P(x≤|T|) = \sum^{min(|T|)}_{x=0} \frac{(\begin{matrix}|S|\\x\end{matrix})(\begin{matrix}|F|-|S|\\|R|-x\end{matrix})}{(\begin{matrix}|F|\\|R|\end{matrix})}\qquad$$
where \(|S|\) means the number of genes in set \(S\). - The \(p_{\_value}(enrichment)\) and \(p_{\_value}(depletion)\) are then corrected by the Bonferroni correction or the false discovery rate (FDR) to represent the true alpha level in the multiple hypotheses testing.
- H3K4ac is said to be enriched/depleted in the promoters of the user's input genes if the corrected \(p_{\_value}(enrichment)\) or corrected \(p_{\_value}(depletion)\) is less than the user-defined threshold.
The procedure for checking whether a specific histone modification is enriched/depleted in the coding regions of the user's input genes is the same as mentioned above except for the definitions of two terms. Now S becomes the set of genes whose coding regions contain the histone modification H3K4ac and \(T=S∩R\) becomes the set of genes whose coding regions contain H3K4ac and are also in the set of the user's input genes.
The procedure for checking whether a specific histone/chromatin regulator (e.g. Esa1) is enriched in the promoters of the user's input genes is the same as mentioned above except for the definitions of two terms. Now S becomes the set of genes whose promoters are bound by Esa1 and \(T=S∩R\) becomes the set of genes whose promoters are bound by Esa1 and are also in the set of the user's input genes.
The web interface of YHMI was constructed using the Python language with the Django MTV framework. The processed histone modification data were deposited in MySQL. All tables, volcano plots and bar charts were produced by the JavaSscript and feature-rich JavaScript libraries (jQuery, DataTables and Plotly.js) to visualize data on the webpage. We also provide the command line version of YHMI (written in Python) for users who want to run YHMI in their local computers (Download command line program here!). YHMI will be maintained by our lab’s research assistants and have two backup sites (http://cosbi4.ee.ncku.edu.tw/YHMI and http://cosib5.ee.ncku.edu.tw/YHMI). Therefore, the long-term stability of YHMI is guaranteed. In the future, we will keep updating YHMI once new histone modification datasets are available in the literature.
Usage of YHMI
YHMI is a web tool for identifying enriched/depleted histone modifications and enriched histone/chromatin regulators in the input gene list. To use YHMI, users have to go through a three-step process.
- Users need to input a list of genes. Standard names, systematic names or aliases are all acceptable.
- Users need to define the sets of genes containing specific histone modifications by choosing the thresholds. For example, by requiring log2(H3K4ac/H3)≥1 (i.e. threshold=1) in the promoters, a set of 1656 genes whose promoters contain H3K4ac could be defined.
- Users have to select the method (Bonferroni correction or FDR) for multiple hypotheses correction and set the p-value threshold (0.01 is used as the default).
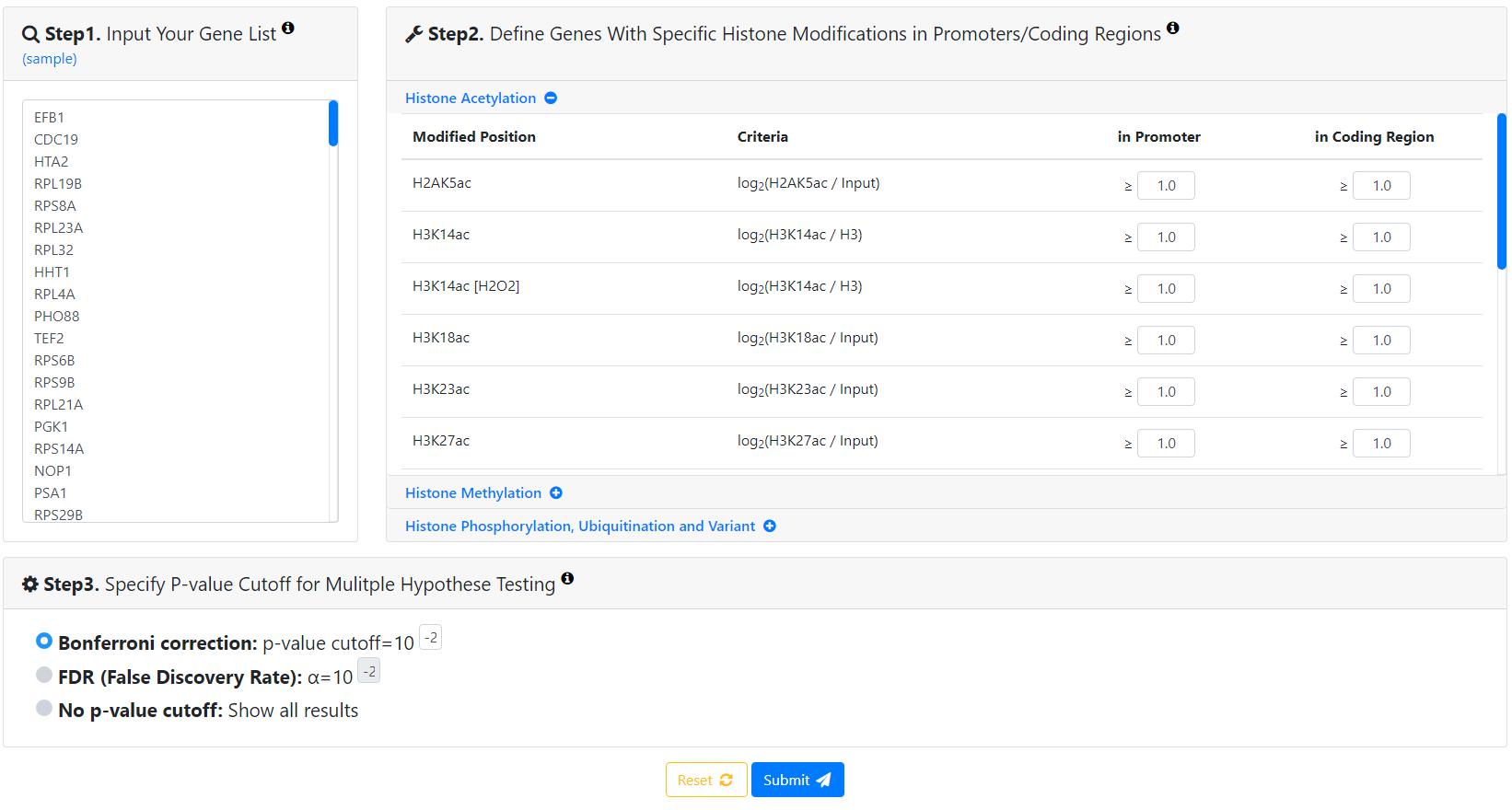
After submission, YHMI will return the identification results which can be divided into two parts.
- The information of the user's settings is shown. Uniquely, we allow users to download all the sets of genes containing specific histone modifications defined by the users for further investigation.
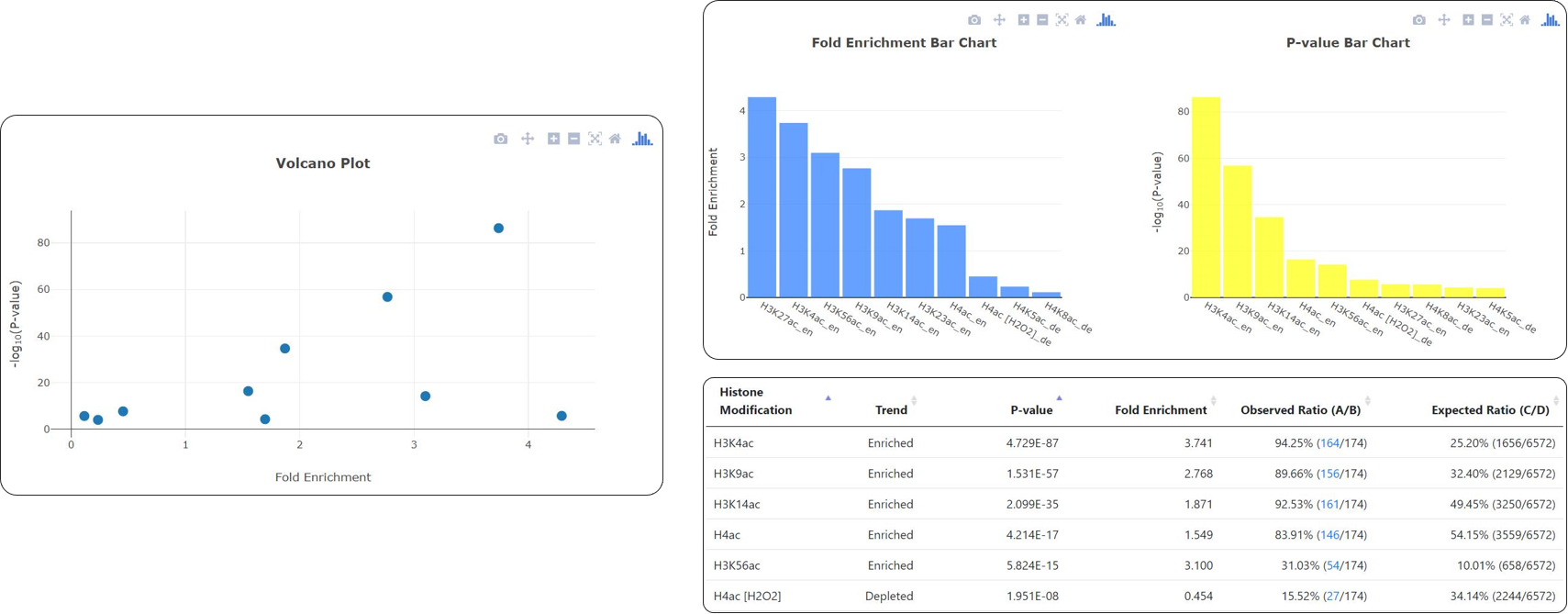
- The identified enriched/depleted histone modifications (acetylation, methylation, ubiquitination and histone variant) in the promoters/coding regions of the input gene list are shown as figures and tables.
Moreover, the enriched histone/chromatin regulators in the promoters of the input gene list are provided.